13310162040
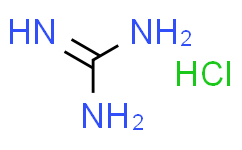
![双[三(羟甲基)氨基甲烷],CAS:6976-37-0](http://struc.chem960.com/strucimg/7000/ctzw0nzi34oyob9fnlmhiwee.png)

单细胞基因组学,表观基因组学,转录组学和蛋白质组学研究揭示出了即使是在同一组织中遗传相同的细胞之间,基因和蛋白质表达依然存在差异。但是来自美国Parker癌症免疫疗法研究所的生物学家Pier Federico Gherardini指出,目前大多数此类研究只检查了每个细胞的单层信息,这可能会产生偏差。“你不能只测量RNA,就由此推测蛋白质看起来都一样。”
现阶段,科学家们已经开始以单细胞分辨率组合多层信息。这些“多组学”技术可以更仔细地观察细胞之间的可变性,更清楚地识别特定细胞及其功能。分析基因组DNA揭示了单细胞基因组,甲基化组织或染色质,而分析RNA和蛋白质则能分别产生转录组和蛋白质组数据。
“多组学比单一层的组学分析更强大,”英国Sanger研究所的分子生物学家Lia Chappell说,“这样才能开始解开所有异质性的真正含义,能够深入挖掘生物机制。”
维也纳CeMM分子医学研究中心的基因组研究员Christoph Bock认为,单细胞多组学特别适用于检查发生快速变化的细胞,如活化的免疫细胞,或非常异质组织中的细胞,包括肿瘤。
这种方法还可以识别在大量群体中被掩盖的罕见但具有生物学重要性的细胞。 Chappell说:“一个典型的例子是那些抗药性低的细胞,在群体研究中我们无法找到它们,因为他们被上千倍更多的细胞淹没了。”
然而,单细胞多组学研究并不容易。目前对于任何单细胞多组学技术,还没有可用的商业试剂盒,并且许多技术还存在限制。研究人员必须修改现有的单细胞方案,使其与多种类型的分子兼容,并且要非常小心地将样品的损失或污染降至zui低。
基因组和转录组
同时对来自相同细胞的DNA和RNA进行测序可以揭示单个细胞之间的基因组变异,解释其转录水平的变化。这样做还可以更准确地检测DNA突变。
DR-seq(DNA-mRNA sequencing)这种测序方法发表于Nature Biotechnology[1],通过裂解单细胞,并同时扩增裂解物中的DNA和RNA。然后将裂解物分成两半,一个用于RNA测序(RNA-seq),另一个用于基因组测序。这样在扩增过程中将DNA和RNA保持在一起,可以zui大限度地减少核酸的损失,但可能会导致潜在的交叉污染。
G&T-seq(genome and transcriptome sequencing)则是另外一种重要方法[2],早在2015年生物通就报道过这种技术(Nature Methods发布重磅测序技术:基因组和转录组平行测序),研究人员当时用G&T-seq对220个小鼠和人类细胞进行测序,获得了空前详细的信息。
具体来说,这种技术就是利用涂有结合mRNA的短寡核苷酸序列的磁珠,物理性地从完全裂解的细胞中分离mRNA和DNA。然后将DNA和mRNA分别扩增和测序。这样让保持mRNA和DNA分离,允许研究人员使用他们选择的方案分析每种分子,但这可能导致核酸的损失。 目前G&T-seq已实现自动化,通量相对较高。
表观基因组和转录组
分析细胞表观基因组和转录组的技术可以揭示甲基化和染色质可接近性(chromatin accessibility,生物通注)对基因表达的调控作用。
“在肿瘤发生等复杂的生物过程中,异质性同时存在于基因组,表观基因组和转录组中,单独分析它们可能不起作用,”北京大学分子生物学家汤富酬教授说。基因相同的肿瘤细胞可能具有不同的DNA甲基化或基因表达模式,可能需要多组学技术才能明确地将它们分类为亚群。
scM&T-seq(simultaneous single-cell methylome and transcriptome sequencing)能同时完成单细胞甲基化组和转录组测序分析[3],这种方法基于G&T-seq,采用相同的方法从单个细胞中分离DNA和RNA,并扩增和测序RNA。对基因组DNA进行亚硫酸氢盐处理,将未甲基化的胞嘧啶转化为尿嘧啶,然后扩增并测序以测定甲基化组。
研发技术人员表示,“这一新的实验方案让你可以并行分析同一单细胞中的DNA甲基化和RNA。我们的方法提供了有关单细胞DNA甲基化异质性与特定基因表达差异之间关系的shou个直观视图。”(Nature Methods:突破性单细胞表观基因组与转录组分析新技术)
scNMT-seq(single-cell nucleosome, methylation, and transcription sequencing)则是在scM和T-seq基础上发展起来的[4],不过这种技术需要将单细胞分离出来,进行处理,检测全基因组染色质可接近性。不同基因组位置的可接近性,或者保护性会影响基因表达,使用scNMT-seq可以发现小鼠胚胎干细胞中表观基因组和转录组之间的新关联。
scMT-seq(another method of simultaneously sequencing single cells’ methylomes and transcriptomes)是同时测序单细胞的甲基化组和转录组的另一种方法[5],这种是加州大学范国平教授和同济大学薛志刚教授研发的,他们利用这种方法对感觉神经元进行研究,揭示了细胞间的转录组和甲基化组异质性。不过,这些差异大多不是启动子甲基化造成的。举例来说,启动子带CpG岛的基因,表达水平与基因体甲基化正相关。这项研究表明,scMT-seq可以用来检测单细胞中的转录组、甲基化组和单核苷酸多态性信息,进而解析表观遗传学基因调控机制。(同济大学发布单细胞测序新技术)
scTrio-seq(single-cell triple omics sequencing是一种单细胞三重组学测序方法[6],由北京大学汤富酬教授等人研发,这种全新的单细胞三重组学测序方法在国际上首次从同一个单细胞中实现对三种组学高通量测序信息的同时获取,并从单细胞水平发现肝癌细胞在三种组学上存在密切相互关联的高度异质性。
scTrio-seq选择性裂解细胞膜,将细胞质中的mRNA与完整细胞核中的基因组DNA分开。对于scMT-seq,实验人员是利用微量移液管收集细胞核,而在scTrio-seq中,则是通过离心分离细胞核。两种情况中,基因组DNA都是进行的改良亚硫酸盐处理和测序方法来检测甲基化组,而来自细胞裂解物的mRNA则是平行扩增和测序。
“基本上在基因组和转录组数据之间没有交叉污染,”汤教授表示。scTrio-seq使用甲基化组序列数据计算评估基因组拷贝数变异,这种技术已用于分析人结直肠癌样本中的异质性。
蛋白质组和转录组
还有几种技术可以同时测定来自单个细胞的转录物和蛋白质。这些方法为科学家提供了转录后的研究,分析导致蛋白质和转录水平之间的差异。
PLAYR(proximity ligation assay for RNA)技术中,蛋白质被不同金属同位素的抗体标记。同时,RNA转录物被同位素标记的探针结合。利用一种称为质谱流式细胞技术(mass cytometry)的方法测量同位素,并且这种技术也可以同时检测每秒数千个单细胞中的40多种不同的mRNA和蛋白质。
CITE-seq(cellular indexing of transcriptomes and epitopes by sequencing)采用寡核苷酸标记的抗体靶向细胞表面蛋白。这种技术分离并裂解单个细胞,并将它们的mRNA和寡标记抗体与涂有短寡核苷酸序列的磁珠结合。扩增RNA和抗体标签并按大小分离,通过测序定量分析蛋白质和转录物。CITE-seq还可同时检测约100种蛋白质以及数万种RNA转录物。
Marlon Stoeckius是纽约基因组中心的分子生物学家,他是这项技术的研发者。目前Stoeckius正致力于将该方法扩展到细胞内蛋白质,不过这需要固定和透化细胞,这可能会降低RNA质量或导致其从细胞中渗出。
REAP-seq(RNA expression and protein sequencing assay)类似于CITE-seq,采用寡核苷酸交联抗体来检测细胞蛋白质和转录物水平(基于测序)。
REAP-seq和CITE-seq都可以检测很多的转录本,但与PLAYR相比,每次检测的细胞数量更少。
“我认为它们是互补的方法,”PLAYR的首席开发人员Gherardini说,“如果你有一个大的队列或临床研究,像PLAYR这样的东西会更加经济有效”。
CITE-seq还有一个优点就是蛋白质定量可以在通常单细胞RNA测序制备中丢弃的部分进行,因此“对RNA测序文库的质量没有任何损害,”纽约基因组中心的Peter Smibert说,“我们认为如果采用RNA-seq作为读数,那么就可以使用CITE-seq。”
如何做出选择
有了这么多种的检测方法可供选择,研究人员还需要根据他们提出的生物学问题,特定技术的成本,劳动密集程度和技术要求来决定使用哪些检测方法。
“通常这需要技术专家团队,计算人员和了解实验系统的生物学家合作才能很好地完成这些类型项目的研究,”Bock说。
如何制定技术方案,选择方法取决于很多因素,各种技术的实验指南存在的一大差异是如何从组织中分离单个细胞,口腔移液和连续稀释是相对快速,简便和低成本的方法,可以zui大限度地降低RNA或蛋白质降解的风险,但它们通量也相对较低。FACS,机器人操作和微流体是高通量的,但它们很昂贵并且可能在细胞分离上没有那么精细。
成本是另一个因素。基于指南和试剂的体积(例如酶和抗体),上述这些技术的价格每个样品从几美元到几百美元不等(这不包括测序的成本,当然测序也可能是限制性的问题)。 “没有多少科学家能够从单个细胞中测序十万个单基因组,”Chappell说。
对于一批样品,单细胞多组学测定可以需要花费超过24小时到近一周的时间,其中涉及手动将细胞核与细胞质分离的方法往往较慢,且劳动强度较大,而基于FACS的方法更方便,通量更高。
生物信息学专业知识是一个优势。 Stoeckius说:“对于所有的这些分析,你需要一点计算机背景和一些R经验,”这是一种用于统计计算和数据分析的编程语言和软件环境。
不过这种情况也可能很快就会改变,因为越来越多的人开始使用这种分析,一些公司开发了易于使用的软件来分析单细胞多组学数据。目前,诸如SEURAT和MOFA之类的计算软件包可以集成来自两个或多个组学层的数据。
随着单细胞单组学技术变得更加灵敏,准确和高通量,相应的多组学技术也可能得到改善。研究人员还致力于将单细胞技术与其它数据层相结合,例如空间信息(如CODEX)和功能分析。将细胞的空间信息链接到其他组学层可以帮助研究人员在组织内映射不同的细胞类型和功能。
zui终的目标还是从单个细胞中捕获所有分子的信息——一种“全方位”的信息,但这仍然需要几年时间。目前,单细胞多组学正在改变科学家处理分子生物学的方式。